Door Gert van Maanen - 16-05-2020 - Microbiologie
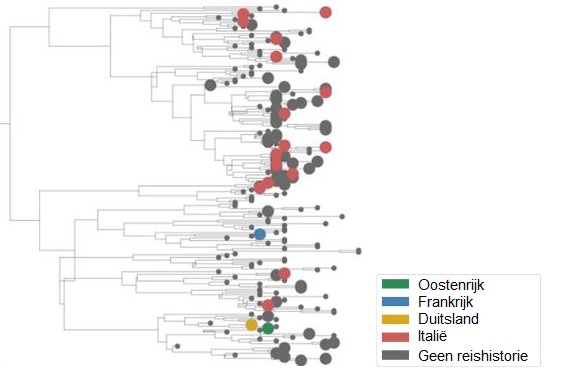
Detail uit de stamboom van Nederlandse sars-cov-2-besmettingen (grote bolletjes) herleid op de reisgeschiedenis. Bron: Oude Munnink e.a., BioRxiv 25 april 2020 (met aanpassingen)
‘Wij hadden ons wél goed voorbereid. Na de eerste meldingen van het nieuwe coronavirus in China in januari hebben we het genetisch volgsysteem voor griepvirussen al klaargezet om gegevens van sars-cov-2 mee te verwerken. Het nieuwe coronavirus is daarmee het tweede virus waarvan we de genetische veranderingen op de voet kunnen volgen’, vertelt viroloog en bioinformaticus Bas Oude Munnink van het Erasmus Medisch Centrum (MC). Hij is eerste auteur van een prepublicatie – onder review voor Nature Medicine – over hoe genoomsequencing van sars-cov-2 de advisering van het Nederlandse Outbreak Management Team heeft ondersteund (BioRxiv, 25 april). ‘Zo is het advies om Noord-Brabant begin maart op slot te doen gebaseerd op de analyse dat er ook zelfstandige besmettingen binnen de provincie plaatvonden en die niet meer uitsluitend te herleiden waren op een reisgeschiedenis in het buitenland’, vertelt Oude Munnink. De eerste twee gedocumenteerde Nederlandse sars-cov-2-besmettingen zijn in Rotterdam binnen twee dagen compleet gesequenst en waren 29 februari beschikbaar. Hiertoe zijn de 30 duizend basisparen van het virus opgedeeld in 83 deels overlappende amplicons van vijfhonderd baseparen, met Nanopore-sequencing geanalyseerd en vervolgens aan de hand van algoritmes gereconstrueerd. ‘Van een monster dat voor 15.00 uur bij ons binnen is, hebben we de avond erna de complete sequentie’, vertelt Oude Munnink trots.
Sars-cov-2 kent volgens hem gemiddeld één mutatie in de anderhalf tot twee weken. In de illustratie rechts zijn de kleine genetische verschillen door zulke mutaties via een zogehete BEAST-analyse – Bayesian evolutionary analysis by sampling – vertaald in een stamboom of fylogenie van de geanalyseerde sars-cov-2-monsters en in tijd gekoppeld aan de reisgeschiedenis van betrokken patiënten. De grote bolletjes zijn Nederlandse gevallen en de kleur betekent dat de besmetting is te herleiden op Oostenrijk (groen), Frankrijk (blauw), Duitsland (geel) of Italië (rood). Voor deze analyse waren er op 15 maart 189 virusgenomen van Nederlandse patiënten gesequenst en ingevoerd in GISAID (Global Initiative on Sharing All Influenza Data). In het begin was Nederland hofleverancier met bijna een kwart van de gegevens, maar inmiddels neemt de Verenigde Staten – met bijna 4.835 van de 17.764 genomen (stand 9 mei) – de toppositie in. Analyses aan GISAID zijn alleen mogelijk voor onderzoekers die kunnen inloggen, maar via de publieke website Nextstrain.org zijn uit delen van de data allerlei selecties en verschillende vormen van stambomen van sars-cov-2 te genereren.
‘Zo is de pandemie bijna in real-time te volgen. Maar je moet je wel steeds realiseren dat landen als Italië en Iran, die waarschijnlijk een grote rol hebben gespeeld in de mondiale verspreiding van het virus, nog maar heel weinig genoomgegevens hebben bijgedragen. Je krijgt al snel een vertekend beeld als je niet weet wat er in die landen is gebeurd’, waarschuwt Oude Munnink. Opmerkelijk is volgens hem wel dat er nog geen enkele Nederlandse besmetting rechtstreeks is terug te leiden op China. ‘Een interessante waarschuwingsfunctie ontstaat als je in een tak van de stamboom ziet dat die geassocieerd is met veel opnames in de intensive care. Want dat kan erop wijzen dat er sprake is van een mutatie die deze virustak extra gevaarlijk maakt. Toe nu toe hebben we dat nog nergens gevonden’, aldus Oude Munnink. Het onderzoeksteam van het Erasmus MC blijft de komende tijd de verspreiding van het virus monitoren om een vinger aan de pols te houden indien er specifieke mutaties optreden. (GvM)
Meer sars-cov-2-nieuws op pagina 14
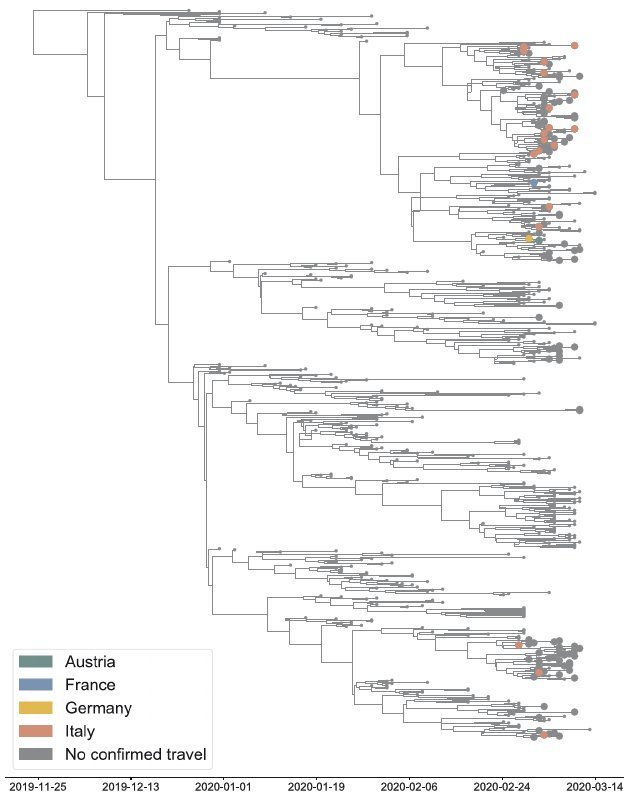
Herkomst van Nederlandse besmettingen met sars-cov-2 (grote bolletjes) herleid op de reisgeschiedenis. Bron: Oude Munnink e.a., BioRxiv 25 april 2020