
Afbeelding van een 3D atomair model van een sars-cov-2-virusdeeltje, op basis van cryo-electrontomografie-scans, machine learning en visuele algoritmen. Foto: Nanographics GMBH, NANOGRAPHICS.AT
In ijltempo proberen onderzoekers de eigenschappen van nieuwe varianten van het coronavirus te begrijpen. Uiteindelijk moet dat leiden tot continue monitoring. ‘Je verwacht dat mutaties één voor één verschijnen, en niet opeens met 22 tegelijk.’
Brits, Zuid-Afrikaans, Braziliaans: de collectie coronavarianten wordt alsmaar groter. En dat zijn dan nog de drie namen die tot nu toe de krant halen. Vorige week werd in Los Angeles bijvoorbeeld een Californische variant (CAL.20C) gesignaleerd met een reeks verdachte mutaties. Nog niet eerder was de wetenschap real time getuige van zo’n wereldomspannend evolutionair proces. Het tempo doet zelfs onderzoekers even achter de oren krabben.
Evolutie
Vanuit de biologie bekeken zijn mutaties niet verrassend, zegt Pleuni Pennings, evolutiebioloog bij San Francisco State University in Californië. ‘Maar ik had niet verwacht dat er nu al zulke grote effecten van evolutie merkbaar zouden zijn, met een virusvariant die 50 procent sneller kan verspreiden.’ Pennings bestudeert evolutie van hiv in patiënten die jarenlang antivirale middelen slikken. ‘Coronavirus en andere verkoudheidsvirussen hebben een veel kortere infectieduur, en daardoor treedt evolutie binnen de patiënt niet zo snel op. Maar door wereldwijde verspreiding kunnen nu toch een groot aantal mutaties optreden.’
Dat laatste heeft alles te maken met moleculaire biologie en blind toeval. Als rna-polymerases van sars-cov-2 in de gastheercel het virusgenoom kopiëren, dan pakken ze soms de verkeerde bouwsteen. De meeste van die vergissingen worden gecorrigeerd, en daardoor zijn mutaties zeldzaam.
Het is nog niet eenvoudig te verklaren welke eigenschappen het succes van de nieuwe varianten bepalen
Maar in deze pandemie speelt ook de wet van de grote getallen. Er lopen dag na dag miljoenen geïnfecteerde mensen rond, dus het virus wordt per etmaal miljarden malen gekopieerd. Rna-polymerases van sars-cov-2 maken zo opgeteld toch wel veel van die zeldzame foutjes. De meeste mutaties hebben geen of nadelige invloed, maar sommige pakken positief uit. Via natuurlijke selectie komt een fitnessvoordeel vanzelf bovendrijven, in de gestage opkomst van nieuwe varianten, die de oude wegconcurreren.
VACCIN
‘Hoe meer geïnfecteerde mensen, hoe groter de kans dat een bepaalde mutatie of combinatie van mutaties de kop opsteekt’, zegt Pennings. Extra zorg levert volgens haar de evolutionaire druk die vaccinatie het komend jaar gaat opleveren. ‘Als de helft van een land gevaccineerd is, en de andere helft is geïnfecteerd, dan komt het virus voortdurend gevaccineerde gastheren tegen. Dan is er meer kans dat een ongevoelige escape -mutant voet aan de grond krijgt. Daarom is het belangrijk dat het aantal besmettingen met het virus zo laag mogelijk blijft en dat iedereen snel het vaccin krijgt.’
Vooral mutaties die de aminozuurvolgorde van het spike-eiwit veranderen trekken de aandacht, omdat die de koppeling met celreceptoren kunnen beïnvloeden. De werking van antistoffen draait om het blokkeren van die interactie. Daarom blijft vaccineren nog wel even nodig, verwacht Pennings: ‘Het virus zal niet uitsterven, want we kunnen niet de hele wereld vaccineren. En het virus zal blijven muteren. Maar m-rna-vaccins kunnen redelijk makkelijk geüpdatet worden, dus ik verwacht dat dat gaat gebeuren.’
Mutatietempo
Het tempo waarmee sars-cov-2 muteert past bij coronavirussen, zegt Bas Oude Munnink, geneticus bij de afdeling viroscience van het Erasmus Medisch Centrum. ‘Net zoals in het begin van de pandemie zien we elke twee weken gemiddeld één van de dertigduizend nucleotiden veranderen.’ Wie de eerst geïsoleerde stam uit Wuhan vergelijkt met een coronavirus dat vandaag in Nederland wordt gevonden, ziet dat gemiddeld vijftig nucleotiden verschillen. Het totaal aantal waargenomen mutaties is overigens veel groter. Alleen al van het gen dat codeert voor het spike-eiwit zijn wereldwijd vierduizend verschillende rna-mutaties bekend. Mutaties leveren gek genoeg een voordeel voor de wetenschap. Het traceren van besmettings-clusters is sinds februari 2019 geleidelijk eenvoudiger geworden, zegt Oude Munnink. ‘Je kunt nu ook steeds meer regionale verschillen detecteren. Zo konden we door de subtiele verschillen tussen varianten dertig of veertig verschillende importgevallen van de Britse variant in Nederland traceren. Dat laat ook zien dat er in december veel is gereisd.’
De Britse variant leek in de loop van januari opeens overal op te duiken: Zuid-Holland, Drenthe, Friesland. ‘De verspreiding in Nederland is nog lastig te duiden, omdat we niet alles kunnen sequensen’, zegt Oude Munnink. ‘In Engeland proberen ze 10 procent van de positieve testen te sequensen. Duitsland gaat 200 miljoen euro in dit soort onderzoek investeren. Ik verwacht dat we in Nederland ook een inhaalslag gaan maken. We zijn al bezig met steekproeven, door materiaal op te vragen van testlabs verspreid door Nederland, maar dat zijn dertig à veertig monsters per lab per maand, dus het is nog een klein percentage.’ Surveillance door sequensen maakt het al vanaf het begin van de pandemie mogelijk om een fylogenetische analyse van coronavarianten te maken.
Nextstrain
Op het open source-platform Nextstrain.org worden bijvoorbeeld dergelijke stambomen gereconstrueerd. Zo kunnen nieuwe waarnemingen en de dynamiek van de pandemie beter worden begrepen.
Het lab van Oude Munnink werkt op dit moment aan het sequensen van monsters uit het bevolkingsonderzoek onder 63 duizend inwoners van de gemeente Lansingerland. Dat project moet een beter beeld geven van het voorkomen van de Britse variant. De resultaten zullen mede bepalen of scholen in februari kunnen heropenen. ‘Het labonderzoek verloopt in drie stappen: eerst de reguliere covid-PCR-test, vervolgens een specifieke PCR die de kenmerkende mutatie van de Britse variant detecteert, en van die gevallen wordt de rna-volgorde bepaald om de hele sequentie in kaart te brengen.’
Corona-landschap
Het is nog niet eenvoudig te verklaren welke eigenschappen het succes van de nieuwe varianten bepalen. Zo is er vorig jaar door veel onderzoeksgroepen lang gepuzzeld op de relatief eenvoudige mutatie D614G, die tussen februari en juni 2020 het wereldwijde corona-landschap ging domineren. Inmiddels hebben epidemiologische analyses, bindingsproeven, dierexperimenten en computersimulaties meer geleerd. Het virus kan met mutatie D614G efficiënter repliceren en verspreiden in het lab. Bij infecties bij hamsters is duidelijk een toegenomen fitness van het gemuteerde virus te zien. Op moleculaire schaal zit de verklaring in de verandering van het aminozuur aspartaat in glycine, waardoor het receptorbindingsdomein van het spike-eiwit wat flexibeler is geworden. De ‘open’ conformatie vergroot mogelijk de kans op succesvolle koppeling met de receptor op de cel. Tegelijkertijd zijn er onderzoekers die concluderen dat de mutatie leidt tot een hogere dichtheid van het spike-eiwit op de virusoppervlak.
SURVEILLANCE
Het onderzoeken van één aminozuur kost tijd. Ingewikkelder wordt het met nieuwe varianten, met twee dozijn rna-mutaties waarvan drie of vier aminozuurveranderingen opleveren in het spike-eiwit. ‘Die nieuwe varianten staan heel ver af van wat tot dan toe bekend was. Het is ook moeilijk te achterhalen wat er precies is gebeurd. Het virus is letterlijk alle kanten op gegaan sinds het begin van 2020. Je verwacht dat mutaties één voor één verschijnen, en niet opeens met 22 tegelijk. Het idee is dat zulke varianten zijn ontstaan in een patiënt met een slecht functionerend immuunsysteem. Zo kon het virus langdurig repliceren en muteren. Een andere mogelijkheid is dat het virus in een gebied heeft gecirculeerd waar weinig tot geen genomische surveillance plaatsvindt.’ Opvallend is dat nieuwe varianten uit Engeland, Zuid-Afrika en Brazilië soms identieke mutaties dragen, zoals K417N, E484K en N501Y. Dat is een duidelijk signaal van convergente evolutie. Als op drie continenten precies dezelfde mutatie komt bovendrijven, dan zou het wel heel toevallig zijn als die geen fitnessvoordeel met zich meebrengt, zegt Oude Munnink.
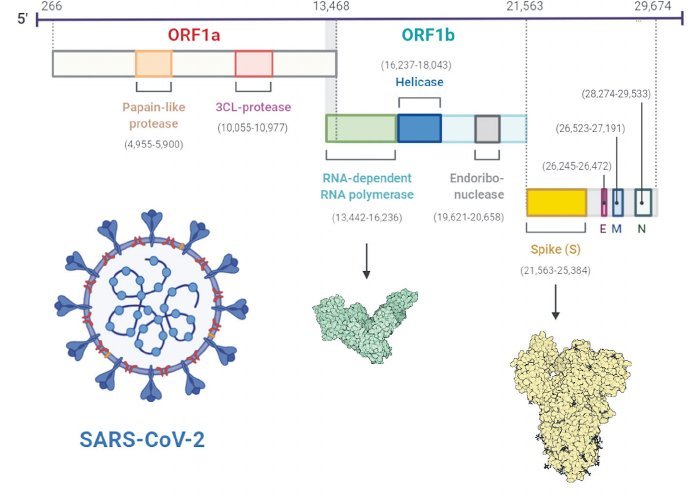
Het genoom van sars-cov-2 telt zo’n 30 duizend letters, die coderen voor diverse enzymen en andere eiwitten, waarvan maar een deel in de virusdeeltjes zijn terug te vinden. Veel onderzoek focust zich op mutaties in het spike-eiwit, maar ook rna-afhankelijke rna-polymerase (RdRp) muteert, wat mogelijk leidt tot hogere mutatiesnelheden. Illustratie: Bewerking van origineel van Tahir Ul Qamar e.a., PLOS ONE 15(12): E0244176
Sequensen levert alleen niet alle antwoorden. Er is net als bij de karakterisering van D614G veel aanvullend labonderzoek nodig. Maar hoe meer men begrijpt, hoe beter men potentieel gevaarlijke mutaties kan herkennen en voorspellen. Mogelijk kan rioolwateronderzoek helpen bij de vroeg-signalering. ‘We kijken dan met extra aandacht naar het receptorbindingsdomein, en de antilichamen die daartegen gericht zijn. Om dat te begrijpen, moet echt nog meer werk worden gedaan.’
Voorspellen en herkennen is al mogelijk bij influenzavirussen, waar op basis van sequentie-analyse hoog-pathogene varianten van het vogelgriepvirus reden zijn om pluimveebedrijven te ruimen. ‘Dat zou je bij coronavirus ook willen ontwikkelen. Dan kun je na signalering van een zorgwekkende variant een stad of regio in lockdown doen.’
Kaders:
Polymerasemutaties
Sars-cov-2 neemt zoals veel virussen zijn eigen kopieermachine mee, in de vorm van een gen dat codeert voor een rna-afhankelijke rna-polymerase (RdRp). Het RdRp -gen kan net als alle andere letters in het 30 duizend letters tellende genoom mutaties oplopen.
Dat levert soms een rna-polymerase dat iets slordiger zijn werk doet. Zo kan in volgende generaties van het virus meer genetische diversiteit ontstaan, en dus meer grondstof voor adaptatie aan de gastheer. Zo’n toename van mutatiefrequentie door selectie op polymerasemutaties is al bekend van bijvoorbeeld hiv. Ook bij sars-cov-2 zijn er inmiddels eerste observaties dat sommige mutaties in RdRp evolutionair voordeel opleveren. Onderzoekers zien dat bepaalde RdRp -mutaties opvallend vaak voorkomen. Bovendien lijken deze mutaties samen te gaan met een iets hogere mutatiefrequentie.
Marters, katten en honden
Bas Oude Munnink publiceerde met collega’s 8 januari in Science over de analyse van corona-uitbraken op Nederlandse nertsenfarms, en de varianten die daarbij de kop opstaken. Op de farms ontstond opvallend snel nieuwe genetische diversiteit, met gemiddeld drie à vier puntmutaties. Waarschijnlijk is dat het gevolg van het grote aantal dieren dat het virus in korte tijd passeerde. ‘We weten niet precies hoeveel dieren zijn geïnfecteerd, dus het is lastig om het exact te kwantificeren. We zagen wel het begin van een herkenbare nertsen-coronalijn ontstaan, maar die hebben we gelukkig alleen bij de dieren en nertsenhouders gevonden, en niet in de algemene populatie. Gastheer-specifieke mutaties baren wel zorgen. Zo kan het virus op de lange termijn ook pathogener worden voor de mens.’
De verspreiding van het virus onder dieren levert een apart evolutionair speelveld. Een dierpopulatie kan een reservoir gaan vormen, waar het virus buiten ons zicht nieuwe mutaties verzamelt. Oude Munnink is betrokken bij een onderzoeksproject dat kijkt naar voorkomen van covid bij wilde dieren in de omgeving van inmiddels geruimde nertsenboerderijen. Bij andere marterachtigen zou het virus ook kunnen rondgaan, zoals steen- en boommarter. Op dit moment wordt dat nader bekeken, net als verspreidingsdynamiek in huisdieren, zoals konijnen, katten en honden.